注:流程工具选择需结合样本类型、基因组注释完整性灵活调整,完整代码参考文献[[4][5]9。
1. 数据质控(Quality Control)
- 目的:剔除低质量序列与接头污染
- 工具:
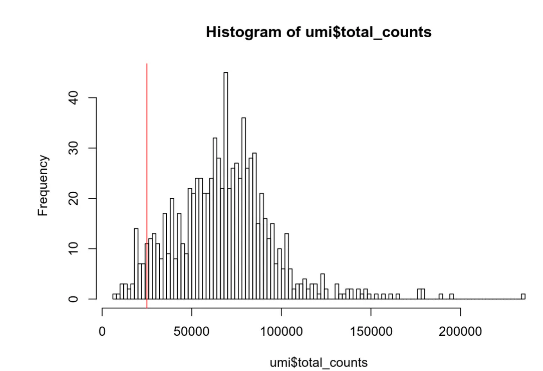
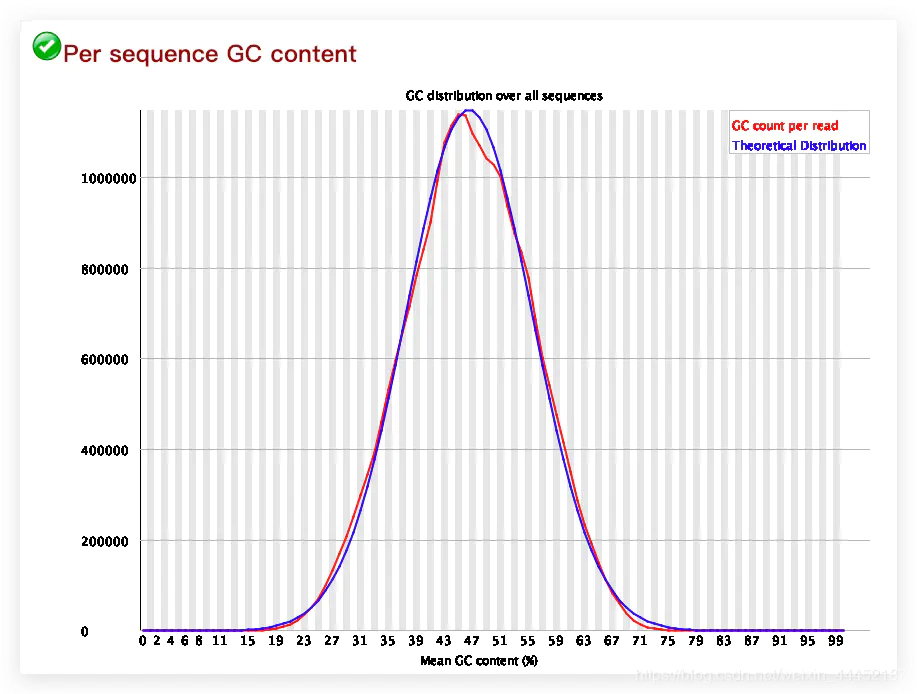
FastQC可视化质量分布,fastp/Trimmomatic过滤低质量碱基(如N碱基占比>5%)、修剪接头[[1][3]7。 - 关键指标:Q30>80%、GC含量稳定、无接头残留✨
2. 序列比对(Sequence Alignment)
- 流程:将质控后reads比对至参考基因组
- 工具:
HISAT2(主流选择,高效省内存)[[2][4]10STAR(适用大基因组,支持可变剪接)[[5]9
- 输出:SAM/BAM格式比对文件,需经
SAMtools排序、去重、索引[[4]12🔥
3. 表达定量(Expression Quantification)
- 基因水平:
StringTie组装转录本并生成FPKM/TPM矩阵[[2][5]10 - 转录本水平:
Salmon/kallisto无比对定量(适合无参考基因组)[[5]9 - 计数矩阵:
prepDE.py转换TPM为raw counts(用于差异分析)[[2]10📊
4. 差异表达分析(DEGs Identification)
- 工具对比:
DESeq2(负二项分布,小样本稳健)edgeR(适用多组学比较)[[1]11
- 筛选标准:|log2FC|≥1 & padj<0.05
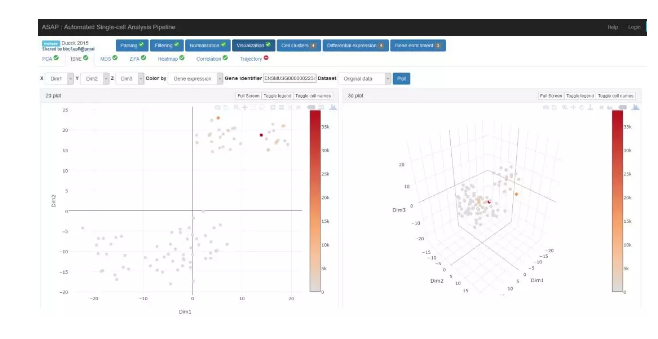
- 可视化:火山图标记显著基因,热图展示聚类模式[[8]11🧬
5. 功能富集与解释(Functional Annotation)
- 基因注释:基于
GO、KEGG数据库解读DEGs功能[[1]6 - 富集分析:
clusterProfiler(R包,支持通路富集)- 缩小焦点基因:筛选与表型强相关基因或关键通路核心基因[[1]6
- 深度挖掘:WGCNA构建共表达网络,锁定核心模块[[6]8🔍
网友热评 💬
@生信小白逆袭中:原来Hisat2+StringTie是黄金组合!实操过酵母数据,流程清晰到哭,Galaxy平台对新手太友好了🥹[[2]10
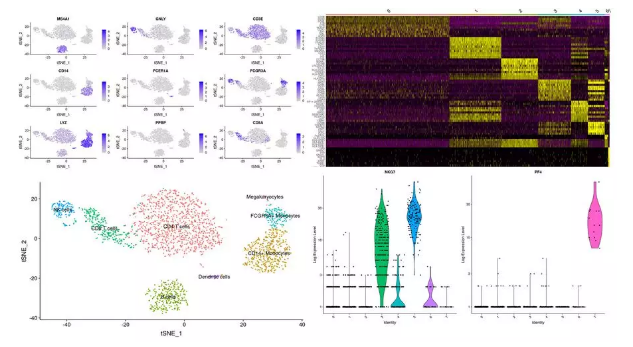
@实验室永动机:被审稿人虐三次才学会:热图别堆全部基因!选Top50 DEGs+关键通路基因,故事感瞬间拉满📈[[8]11
@科研猫奴:差异基因筛完后一定要做PPI网络啊!上周筛到个冷门转录因子,连上免疫通路直接补了机制缺口~[[6]8
转录组数据分析全流程解析 💻
基于最新文献与实操指南整理,覆盖从原始数据到生物学解释的核心步骤👇
相关问答
转录组测序的数据分析流程是怎样的? 答: 转录组测序的数据分析流程主要包括以下几个关键步骤:
原始数据质量控制:目的:确保测序数据的可靠性和准确性,为后续分析打下坚实基础。操作:使用生物信息学工具对原始测序数据进行质量评估,包括检查碱基质量分布、测序深度、测序错误率等指标。序列比对:目的:将测序得到的序列与参考基因组或转录组进行匹配...
核苷酸分析 企业回答:咨询上海兆维,上海兆维科技发展有限公司是一家致力于核苷、核苷酸、修饰性核苷、基因单体、靶向示踪剂、生物酶等产品研发、规模化生产的企业,公司的产品主要应用于核酸药物、mRNA 疫苗/药物、基因测序、分子诊断等领域。作为国内外核酸药物和 ... 转录组学分析的流程? 答:下面是一般的转录组学分析流程:样品收集与处理:首先,从感兴趣的生物体细胞或组织中收集样品,注意采集要符合生物安全和伦理规范。然后,对样品进行预处理,包括细胞破碎、RNA提取和纯化等步骤。RNA测量和质检:通过分光光度计或荧光探针等方法,测量样品中的总RNA浓度,并进行质量评估,确保RNA质量良好,...
文章来源: 用户投稿版权声明:除非特别标注,否则均为本站原创文章,转载时请以链接形式注明文章出处。





